Research
Alzheimer’s Disease (AD)
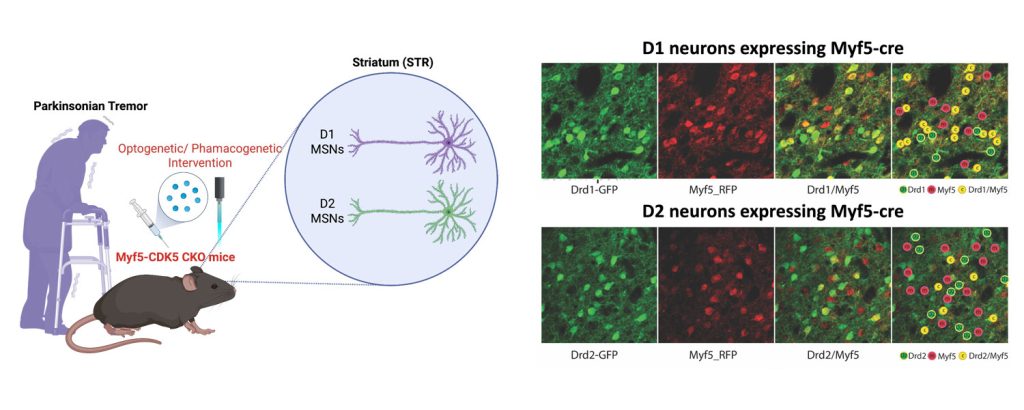
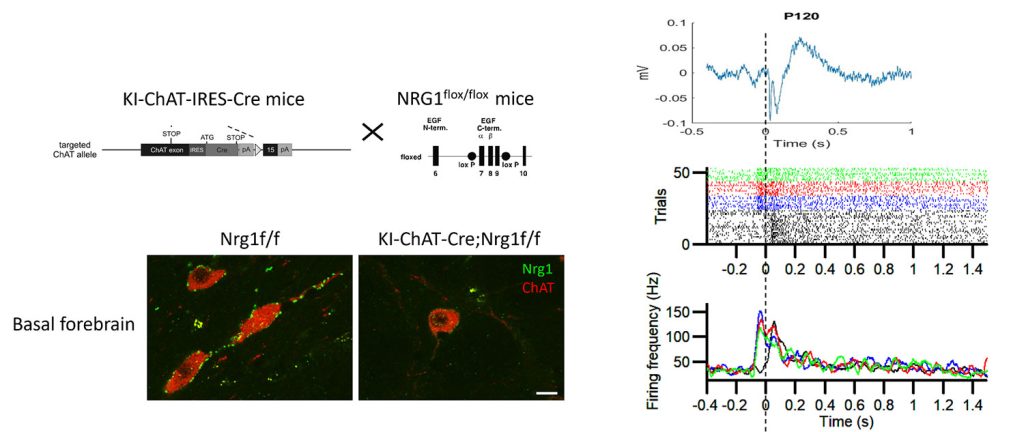
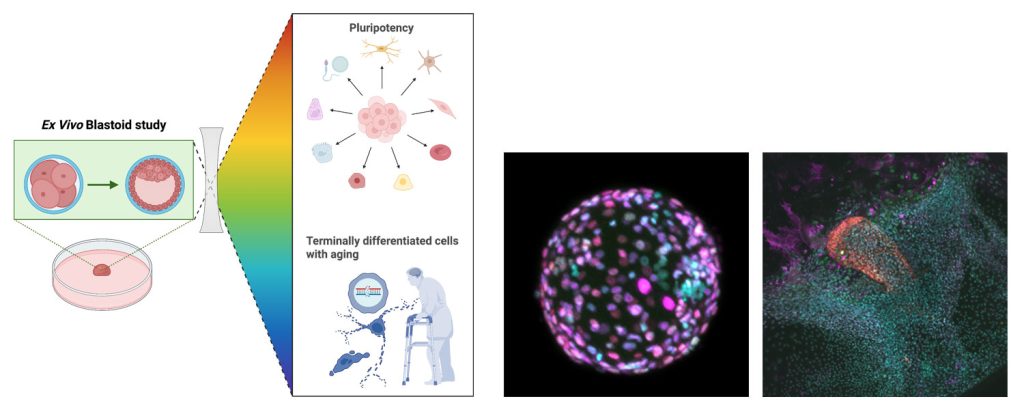
Circuit-specific vulnerable cell types and signaling pathways in aging and AD
Investigators: Xinji Wang, Evan Booker, Bertha Dominguez, Ling Zhang, Eleanor Ketterer-Sykes, and Hanhee Jo
We view that the brain is geometrically organized by interconnected hubs or communities, comprising both specific neuronal and non-neuronal cells that function similar to domestic or global airport hubs. This project aims to understand what and how certain brain hubs and their cell types and circuits are more vulnerable to initiate and promote progression of Alzheimer’s disease (AD) in response to proteinopathies (e.g., tau tangles and amyloid-? plaques). AD shows striking age- and sex-dependent patterns of progression. Several lines of evidence suggest that AD is initiated in subcortical brain structures, including the locus coeruleus, hypothalamus, amygdala, and entorhinal cortex, before spreading to the hippocampus and neocortex.
Although AD risk genes are broadly expressed, accumulating evidence suggests that vulnerability depends on the interplay between neurons and glial cells and their differential responses to protein stress. As aging is one of major risk factors in AD and is regulated by longevity genes, we will investigate the interplay of longevity and AD gene networks, as well as the influence of environmental factors and lifestyles during the progression of aging and AD.
Using genetically engineered humanized AD mouse knock-in models (e.g., MAPT, APP, APOE2/3/4, and TREM2), we combine barcoded single neuronal projection mapping and single-nucleus multi-omics with spatial transcriptomics to identify circuit-specific vulnerable versus resilient cell types across disease stages. AI-enabled behavioral tests are developed to unbiasedly and longitudinally identify deficits that may be underpinned by circuit-specific alterations and inform early detection of AD. By integrating behavioral and multimodal single-cell datasets, we aim to uncover the circuit-specific mechanisms driving selective vulnerability in AD at the predementia phase and reveal new molecular pathways (e.g., mitochondrial and metabolic impairments, and neuroinflammation), that may in turn inform therapeutic strategies.
Importantly, we will leverage datasets with large-scale drug-induced transcriptomic signatures and electronic medical records to identify candidate drugs to probe disease mechanisms and translate into clinically relevant contexts. Integrating genomic and proteomic profiles from AD cohorts with our experimental results will help prioritize druggable targets and accelerate drug discovery for early intervention. Finally, we have developed and will continue to develop a suite of enhancer-based viral vectors (e.g., cell- and sex-specific) for non-invasive delivery (e.g., focus-ultrasound sonication) of therapeutic cargos.
Mechanisms underlying selective AD vulnerability in females
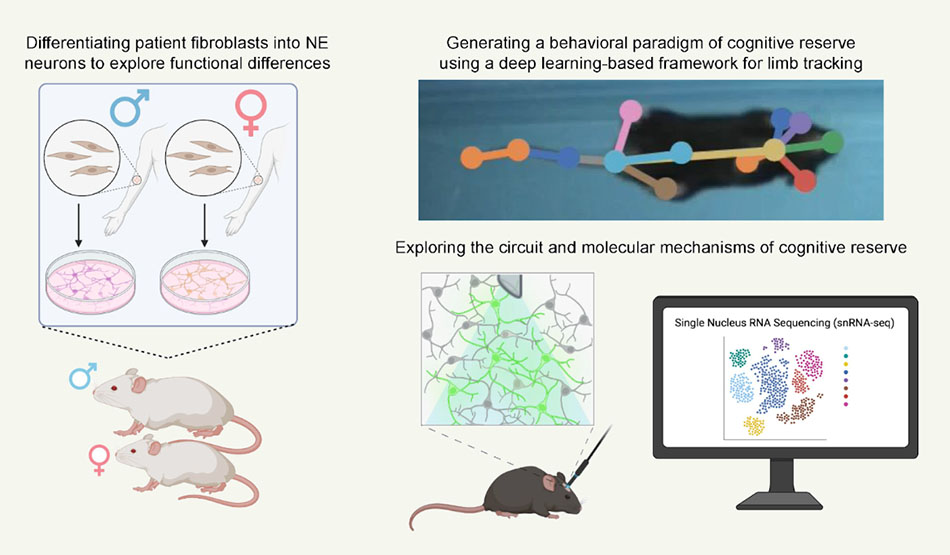
Investigators: Eleanor Ketterer-Sykes
Women are two- to three-times more likely than men to develop Alzheimer’s-related dementia and have a higher likelihood of exhibiting pathologies of greater severity and rapid progression. However, female subjects are a historically understudied demographic in scientific research, meaning we have little understanding of the mechanisms underlying this selective vulnerability. We aim to explore how demographics are differentially impacted during the early stages of the disease. For example, we will elucidate the cell-type specific circuit and molecular mechanisms underlying cognitive reserve, or the capacity to maintain cognitive abilities despite neuropathophysiological changes associated with aging or neurodegenerative disease, which has been proposed to contribute to this selective resiliency versus vulnerability.
Alternative splicing and gene isoform alterations in AD

Investigators: Hanhee Jo and Ling Zhang
Alternative splicing expands transcriptomic and proteomic diversity by allowing a single gene to generate multiple isoforms with distinct functions. In the brain, where splicing complexity is especially high, dysregulation of isoform usage is increasingly recognized as a contributor to neurodegenerative diseases, including AD. For example, splicing of the MAPT gene generates tau isoforms with three or four microtubule-binding repeats, and imbalances in their ratios have been directly linked to tau pathology. Similarly, alternative splicing of ApoER2 regulates synaptic plasticity through modulation of the Reelin signaling pathway and has been implicated in AD-related cognitive decline.
Our multi-omics analyses showed a distinctive expression pattern of CDH18, a type II calcium-dependent cell adhesion molecule expressed mainly in the brain. CDH18 orchestrates cell–cell interactions, and our findings suggest it may have an important role in glial–neuronal communication and selective vulnerability in AD. In line with the concept that isoform alterations shape disease mechanisms in AD, we aim to determine how different CDH18 isoforms, both known and novel, influence AD pathology.
Conventional short-read RNA sequencing captures expression levels but often fails to resolve full-length isoforms, leaving critical questions about splice variant dynamics unanswered. Therefore, we leverage single cell long-read sequencing technology to fully capture transcript structures, uncover novel isoforms, identify disease-associated splicing events, and map how isoform usage changes across brain regions, disease stages, and cell types. These insights can reveal underappreciated regulatory mechanisms underlying synaptic dysfunction, glial activation, and neuronal vulnerability in AD, offering new entry points for therapeutic intervention.
Spatial proteogenomic atlas of Alzheimer’s Disease
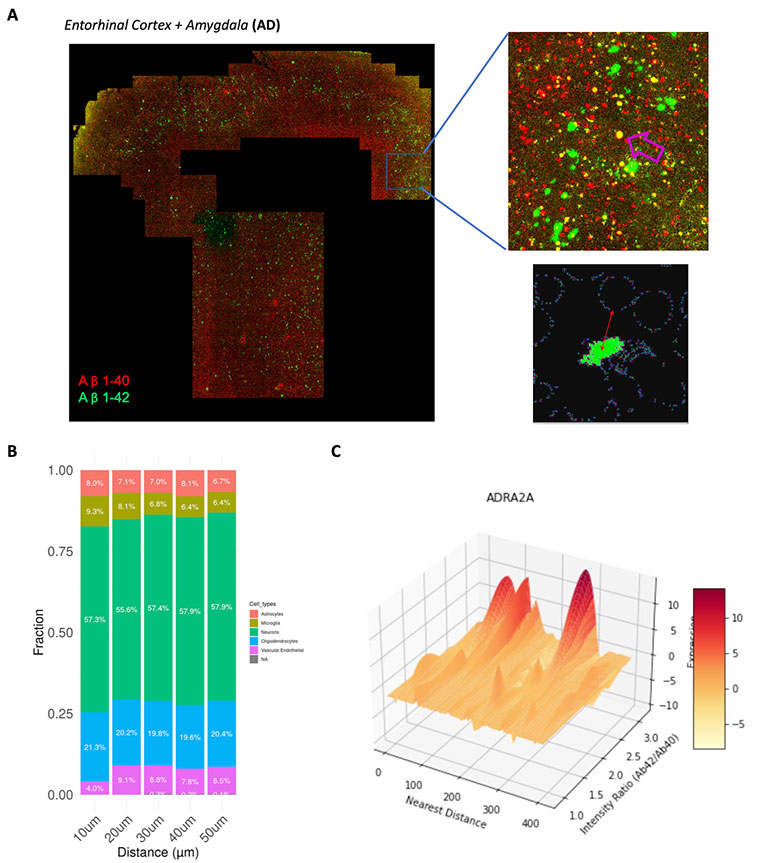
Investigators: Ling Zhang, Evan Booker, and Cheng Ta Lee
Alzheimer’s disease (AD) follows stereotyped neural circuits and accumulates amyloid-? (A?) plaques and tau tangles, yet the spatial rules behind this selective vulnerability remain unclear. We will build a human brain spatial proteogenomic atlas that measures ~6,000 RNAs and 68 proteins on the same FFPE sections from the entorhinal cortex (EC), amygdala, and locus coeruleus (LC). Using protein-guided cell segmentation and joint RNA–protein profiling, we define 20+ in situ cell/state clusters and relate them to pathology by mapping A? plaques (quantified by A?42/A?40) and the local burden of hyperphosphorylated tau (p-tau) as a molecular readout of tau tangles. Plaque- and tangle-centric analyses model distance-dependent shifts in cellular composition and molecular programs, revealing niche-specific responses rather than uniform tissue effects. Regionally, the LC shows reduced noradrenergic neurons with increased p-tau/tangle burden, consistent with early LC involvement. But in the EC, we identify a spatial co-expression module (e.g., RTN1–MEG3) concentrated near plaques/tangles but not in the amygdala—nominating a region-selective vulnerability pathway. Expected outcomes include a cell-resolved map of AD pathology across EC, amygdala, and LC; plaque/tangle distance signatures of glial and neuronal states; and prioritized, region-specific modules for orthogonal validation (RNAscope/IHC) and cross-dataset replication—actionable hypotheses for why certain brain communities succumb first and how to target them therapeutically.